
بیماری آبتالیپوپروتئینمی (Abetalipoproteinemia)
آبتالیپوپروتئینمی (Abetalipoproteinemia)
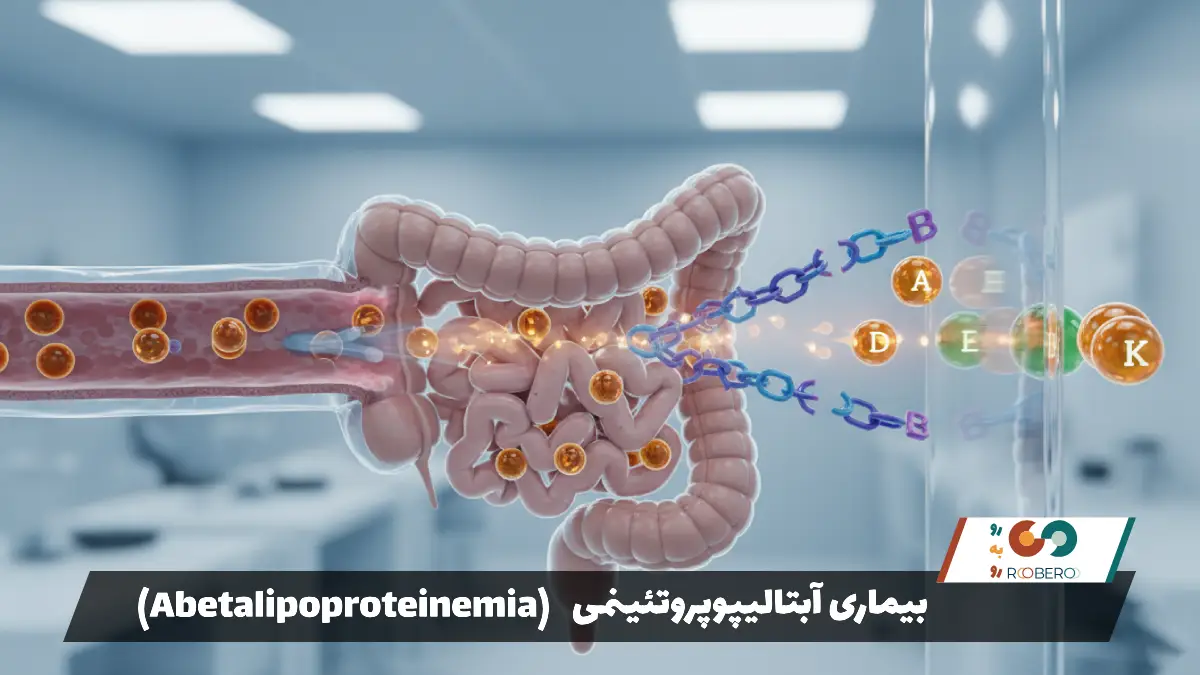
آبتالیپوپروتئینمی، که گاهی با نام سندرم باسن-کورنزوایگ (Bassen-Kornzweig) نیز شناخته میشود، یک اختلال ژنتیکی نادر و ارثی است که بر نحوه جذب و انتقال چربیها و ویتامینهای محلول در چربی در بدن تأثیر میگذارد. بدن انسان برای عملکردهای حیاتی خود، از جمله ساخت سلولها، تولید هورمونها و تأمین انرژی، به چربیها نیاز دارد. چربیهای رژیم غذایی در روده جذب میشوند و سپس باید توسط پروتئینهای خاصی بستهبندی شده تا بتوانند در جریان خون حرکت کنند، زیرا چربی به تنهایی در آب (خون) حل نمیشود. این بستههای حملکننده چربی، لیپوپروتئین نامیده میشوند. در افراد مبتلا به آبتالیپوپروتئینمی، بدن فاقد توانایی تولید لیپوپروتئینهای حاوی آپولیپوپروتئین B (مانند شیلومیکرونها، VLDL و LDL) است.
نتیجه این ناتوانی، اختلال شدید در جذب چربیهای غذایی از روده و انتقال آنها به کبد و سایر بافتها است. این بدان معناست که حتی اگر فرد غذای کافی و پرچرب بخورد، بدنش نمیتواند از آن استفاده کند و چربیها همراه با مدفوع دفع میشوند. علاوه بر چربی، جذب ویتامینهای محلول در چربی یعنی ویتامینهای A، D، E و K نیز به شدت مختل میشود. کمبود شدید این ویتامینها، بهویژه ویتامین E، مسئول بسیاری از عوارض طولانیمدت و مخرب این بیماری بر سیستم عصبی و بینایی است.
این بیماری معمولاً در دوران نوزادی با علائم گوارشی آشکار میشود، اما اگر تشخیص داده نشود، در اواخر کودکی و نوجوانی با مشکلات جدی عصبی و چشمی پیشرفت میکند. شناخت دقیق مکانیسم این بیماری به درک بهتر اهمیت چربیها در بدن کمک میکند. آبتالیپوپروتئینمی نمونهای کلاسیک از بیماریهای متابولیک است که در آن یک نقص مولکولی کوچک منجر به عوارض سیستمیک گسترده میشود. در این مقاله جامع، تمامی جنبههای این بیماری نادر، از ژنتیک و تشخیص تا مدیریت و زندگی با آن، با جزئیات کامل بررسی خواهد شد.
پیشگیری از آبتالیپوپروتئینمی
از آنجایی که آبتالیپوپروتئینمی یک بیماری ژنتیکی با الگوی وراثت اتوزومال مغلوب است، پیشگیری به معنای رایج آن (مانند واکسیناسیون یا رعایت بهداشت برای جلوگیری از بیماریهای عفونی) در مورد آن صدق نمیکند. شما نمیتوانید با تغییر سبک زندگی، رژیم غذایی یا ورزش از بروز جهش ژنتیکی جلوگیری کنید. با این حال، مفهوم پیشگیری در اینجا بر جلوگیری از تولد نوزاد مبتلا در خانوادههایی که سابقه این بیماری را دارند، یا جلوگیری از بروز عوارض شدید در فرد مبتلا متمرکز است.

مهمترین ابزار برای پیشگیری از تولد فرزند مبتلا، مشاوره ژنتیک است. این بیماری زمانی بروز میکند که هر دو والدین ناقل ژن معیوب باشند. والدین ناقل معمولاً هیچ علامتی ندارند و از وضعیت ژنتیکی خود بیخبرند تا زمانی که صاحب فرزندی مبتلا شوند. اگر زوجی سابقه خانوادگی ابتلا به این بیماری را دارند یا ازدواج فامیلی انجام دادهاند (که ریسک بیماریهای مغلوب را افزایش میدهد)، انجام آزمایشهای ژنتیک قبل از بارداری ضروری است. در این آزمایشها، وضعیت ژن MTTP در والدین بررسی میشود. اگر هر دو والد ناقل باشند، در هر بارداری ۲۵ درصد احتمال تولد فرزند بیمار وجود دارد.
در صورتی که مشخص شود زوجین هر دو ناقل ژن معیوب هستند، روشهای پیشرفته کمکباروری مانند تشخیص ژنتیکی پیش از لانهگزینی (PGD) میتواند به عنوان یک روش پیشگیرانه مورد استفاده قرار گیرد. در این روش، لقاح در محیط آزمایشگاه (IVF) انجام میشود و جنینها در مرحله چند سلولی از نظر ژنتیکی بررسی میشوند. تنها جنینهایی که فاقد دو نسخه از ژن معیوب هستند، برای انتقال به رحم مادر انتخاب میشوند. این فرآیند تضمین میکند که نوزاد متولد شده به آبتالیپوپروتئینمی مبتلا نخواهد بود. این تکنولوژی اگرچه پرهزینه و پیچیده است، اما برای خانوادههایی که نگران انتقال بیماری به نسل بعد هستند، گزینهای بسیار ارزشمند محسوب میشود.
جنبه دیگر پیشگیری، “پیشگیری ثانویه” است که به معنای جلوگیری از بروز علائم شدید و ناتوانکننده در فردی است که با این بیماری متولد شده است. تشخیص زودهنگام در دوران نوزادی، کلید اصلی این نوع پیشگیری است. اگر درمان با ویتامینها و اصلاح رژیم غذایی بلافاصله پس از تولد یا در ماههای اول زندگی آغاز شود، میتوان از بسیاری از عوارض عصبی و چشمی غیرقابل بازگشت (مانند نابینایی و ناتوانی حرکتی) جلوگیری کرد یا بروز آنها را به تعویق انداخت. بنابراین، آگاهی پزشکان و والدین نسبت به علائم اولیه گوارشی و عدم وزنگیری نوزاد، نقش حیاتی در پیشگیری از وخامت بیماری دارد.
روشهای درمان آبتالیپوپروتئینمی
درمان آبتالیپوپروتئینمی در حال حاضر درمان قطعی یا شفابخش (Curative) نیست، بلکه درمانی حمایتی و مدیریتی است که باید تا پایان عمر ادامه یابد. هدف اصلی درمان، تأمین مواد مغذی ضروری که بدن قادر به جذب آنها نیست و کنترل علائم گوارشی ناشی از عدم تحمل چربی است. سنگ بنای درمان این بیماری، استفاده از دوزهای بسیار بالای مکملهای ویتامینی است. از آنجایی که مکانیسم جذب طبیعی این ویتامینها از کار افتاده است، پزشکان با تجویز مقادیر بسیار زیاد (مگادوز)، تلاش میکنند تا از طریق انتشار غیرفعال، مقداری از این ویتامینها را به جریان خون برسانند.
ویتامین E مهمترین بخش درمان دارویی است. کمبود این ویتامین عامل اصلی تخریب سیستم عصبی و شبکیه چشم در این بیماران است. دوزهای تجویزی ویتامین E برای این بیماران دهها برابر بیشتر از نیاز روزانه یک فرد عادی است. همچنین مکملهای ویتامین A برای حفظ بینایی و ویتامین K برای تضمین انعقاد خون طبیعی و جلوگیری از خونریزی ضروری هستند. ویتامین D نیز برای سلامت استخوانها و جلوگیری از راشیتیسم تجویز میشود. این مکملها معمولاً به صورت خوراکی مصرف میشوند، اما در موارد شدید ممکن است نیاز به تزریق عضلانی باشد تا جذب آنها تضمین شود. پایش مداوم سطح خونی این ویتامینها برای تنظیم دوز دارو بسیار حیاتی است.
علاوه بر ویتامینها، مدیریت رژیم غذایی بخش جداییناپذیر درمان است. بیماران باید از رژیم غذایی بسیار کمچرب پیروی کنند تا از علائم آزاردهنده گوارشی مانند اسهال چرب، نفخ و درد شکم جلوگیری کنند. با این حال، حذف چربی به معنای حذف کالری است که میتواند منجر به لاغری مفرط شود. برای جبران این مشکل، از نوع خاصی از روغنها به نام تریگلیسیرید با زنجیره متوسط (MCT) استفاده میشود. روغن MCT نیازی به بستهبندی در شیلومیکرونها ندارد و مستقیماً از روده جذب و وارد کبد میشود، بنابراین منبع انرژی مناسبی برای این بیماران است بدون اینکه باعث مشکلات گوارشی شود.
درمانهای حمایتی فیزیکی نیز برای بیمارانی که دچار عوارض عصبی شدهاند، ضروری است. فیزیوتراپی و کاردرمانی به حفظ قدرت عضلانی، بهبود تعادل و افزایش استقلال بیمار در فعالیتهای روزمره کمک میکند. برای مشکلات بینایی، معاینات منظم چشمپزشکی و استفاده از ابزارهای کمکی بینایی توصیه میشود. در موارد پیشرفته که انحراف ستون فقرات یا مشکلات ارتوپدی ایجاد شده است، مداخلات جراحی ممکن است لازم باشد. درمان موفقیتآمیز نیازمند همکاری تیمی از متخصصان شامل متخصص گوارش، متخصص تغذیه، متخصص مغز و اعصاب، چشمپزشک و متخصص ژنتیک است.
نحوه تشخیص آبتالیپوپروتئینمی
تشخیص آبتالیپوپروتئینمی اغلب با چالش روبروست، زیرا علائم اولیه آن میتواند با سایر بیماریهای گوارشی یا سوءجذب اشتباه گرفته شود. فرآیند تشخیص معمولاً با شرح حال بالینی دقیق و معاینه فیزیکی آغاز میشود. پزشک ممکن است با مشاهده نوزادی که دچار اسهال مزمن، عدم وزنگیری مناسب (Failure to Thrive) و مدفوع چرب و بدبو است، به اختلالات سوءجذب مشکوک شود. اگر این علائم با مشکلات عصبی یا تأخیر تکاملی همراه باشد، شک به بیماریهای متابولیک نادر تقویت میشود.
یکی از کلیدیترین و سادهترین روشهای غربالگری و تشخیص اولیه، انجام آزمایش خون کامل (CBC) و بررسی لام خون محیطی (Blood Smear) زیر میکروسکوپ است. در افراد مبتلا به آبتالیپوپروتئینمی، گلبولهای قرمز خون شکل طبیعی و دیسکمانند خود را از دست میدهند و ظاهری خاردار یا ستارهای شکل پیدا میکنند. این سلولها آکانتوسیت (Acanthocyte) نامیده میشوند و وجود درصد بالایی از آنها (معمولاً بیش از ۵۰ درصد) یک نشانه بسیار قوی برای این بیماری است. این تغییر شکل ناشی از ترکیب غیرطبیعی چربیها در غشای سلولهای خونی است.
گام بعدی و تأییدکننده تشخیص، بررسی پروفایل لیپیدی خون (چربی خون) است. در این بیماران، سطح کلسترول کل، تریگلیسیرید و LDL (کلسترول بد) به طرز غیرعادی پایین است. در واقع، سطح کلسترول LDL اغلب غیرقابل اندازهگیری یا نزدیک به صفر است. همچنین سطح پروتئینی به نام آپولیپوپروتئین B (ApoB) در خون به شدت کاهش یافته یا اصلا وجود ندارد. ترکیب “آکانتوسیتوز” در اسمیر خون و “هیپولیپیدمی شدید” (چربی خون بسیار پایین) تقریباً تشخیص را قطعی میکند.
برای تأیید نهایی و دقیق، آزمایش ژنتیک مولکولی انجام میشود. در این آزمایش، نمونه خون بیمار برای یافتن جهش در ژن MTTP بررسی میشود. شناسایی جهش ژنتیکی نه تنها تشخیص را ۱۰۰ درصد تأیید میکند، بلکه امکان بررسی سایر اعضای خانواده و تشخیص ناقلین را نیز فراهم میآورد. سایر آزمایشهای تکمیلی شامل معاینه چشم برای بررسی شبکیه (رتینیت پیگمنتوزا)، الکترومیوگرافی (EMG) برای بررسی سلامت عصب و عضله، و تصویربرداری MRI مغز برای ارزیابی تحلیل مخچه ممکن است انجام شود. همچنین آزمایش مدفوع برای اندازهگیری میزان دفع چربی نیز میتواند شدت سوءجذب را نشان دهد.
نشانههای بیماری آبتالیپوپروتئینمی
نشانههای آبتالیپوپروتئینمی طیف وسیعی از سیستمهای بدن را درگیر میکند و معمولاً در سه مرحله یا دسته اصلی طبقهبندی میشوند: علائم گوارشی، علائم خونی و علائم عصبی-چشمی. اولین علائم معمولاً در دوران شیرخوارگی و پس از شروع تغذیه با شیر خشک یا غذاهای حاوی چربی ظاهر میشوند. نوزاد ممکن است دچار اسهال مزمن، استفراغ، نفخ شکم و دفع مدفوع حجیم، کمرنگ و بسیار بدبو شود که ناشی از چربی هضم نشده است (استئاتوره). این مشکلات گوارشی منجر به عدم رشد کافی، کمبود وزن و سوءتغذیه شدید میشود که اغلب والدین را نگران کرده و آنها را به سمت پیگیری پزشکی سوق میدهد.

با گذشت زمان و در سنین کودکی (معمولاً قبل از ۱۰ سالگی)، در صورتی که درمان مناسب آغاز نشود، علائم عصبی پدیدار میشوند. این علائم ناشی از کمبود شدید و مزمن ویتامین E در بافتهای عصبی است. یکی از اولین نشانهها، فقدان رفلکسهای تاندونی عمقی (مانند رفلکس زانو) است. کودک ممکن است دچار عدم تعادل و هماهنگی در راه رفتن (آتاکسی) شود، بهطوری که تلوتلو میخورد یا مکرراً زمین میخورد. ضعف عضلانی پیشرونده، لرزش دستها و اختلال در حس لامسه و حس موقعیت مفاصل نیز از دیگر علائم نورولوژیک هستند. تکلم ممکن است دچار اختلال شود و در موارد شدید و درماننشده، بیمار ممکن است توانایی راه رفتن مستقل را از دست بدهد.
علائم چشمی معمولاً در اواخر دهه اول یا نوجوانی بروز میکنند. شایعترین مشکل، رتینیت پیگمنتوزا (Retinitis Pigmentosa) است که یک بیماری پیشرونده شبکیه چشم میباشد. علائم اولیه شامل شبکوری و کاهش دید در نور کم است. به تدریج میدان دید محدود شده (دید تونلی) و حدت بینایی کاهش مییابد که در صورت عدم درمان میتواند به نابینایی کامل منجر شود. حرکات غیرطبیعی و پرشی چشم (نیستاگموس) و افتادگی پلک نیز ممکن است دیده شود. علاوه بر این موارد، کمبود ویتامین K میتواند باعث خونریزیهای خودبهخودی یا کبودی آسان شود و کمبود ویتامین D میتواند منجر به نرمی استخوان و بدشکلیهای اسکلتی نظیر انحنای ستون فقرات (کیفواسکولیوز) و قوس زیاد کف پا گردد.
اسمهای دیگر بیماری آبتالیپوپروتئینمی
این بیماری در متون پزشکی و علمی با چندین نام مختلف شناخته میشود که آگاهی از آنها برای جستجو در منابع علمی یا درک گزارشهای پزشکی مفید است. رایجترین نام علمی آن همان “آبتالیپوپروتئینمی” (Abetalipoproteinemia) است که مستقیماً به فقدان لیپوپروتئینهای بتا در خون اشاره دارد. در این واژه، “آ” به معنای فقدان، “بتا” به نوع لیپوپروتئین و “لیپوپروتئینمی” به وضعیت پروتئینهای چربی در خون اشاره میکند.
یکی از مشهورترین نامهای دیگر این بیماری، سندرم باسن-کورنزوایگ (Bassen-Kornzweig syndrome) است. این نامگذاری به احترام دو پزشک به نامهای فرانک باسن (Frank Bassen) و آبراهام کورنزوایگ (Abraham Kornzweig) انجام شده است که برای اولین بار در سال ۱۹۵۰ این بیماری را توصیف کردند. آنها متوجه ارتباط بین گلبولهای قرمز خاردار (آکانتوسیتها)، مشکلات عصبی و رتینیت پیگمنتوزا در یک دختر ۱۸ ساله شدند و این سندرم را معرفی کردند.
علاوه بر این، گاهی اوقات از نامهای توصیفی مبتنی بر نقص بیوشیمیایی یا ژنتیکی بیماری استفاده میشود. عناوینی مانند “کمبود ترانسفراز میکروزومال تریگلیسیرید” (Microsomal Triglyceride Transfer Protein Deficiency) یا به اختصار “کمبود MTP” نیز به کار میروند که دقیقاً به پروتئین معیوب در این بیماری اشاره دارند. همچنین ممکن است با نام “فقدان بتالیپوپروتئین ارثی” نیز در برخی متون قدیمیتر دیده شود. تمامی این اسامی به یک وضعیت واحد اشاره دارند و تفاوت تنها در نحوه نامگذاری (بر اساس کاشف، علائم بالینی یا نقص مولکولی) است.
تفاوت بیماری آبتالیپوپروتئینمی در مردان و زنان
آبتالیپوپروتئینمی از الگوی وراثت “اتوزومال مغلوب” پیروی میکند. کروموزومهای اتوزوم، کروموزومهای غیرجنسی هستند که در مردان و زنان یکسان میباشند. ژن MTTP که مسئول این بیماری است، بر روی کروموزوم شماره ۴ قرار دارد. این بدان معناست که جنسیت فرد هیچ نقشی در احتمال به ارث بردن ژن معیوب ندارد. بنابراین، شیوع این بیماری در مردان و زنان تقریباً برابر است و هیچ یک از دو جنس استعداد ژنتیکی بیشتری برای ابتلا ندارند.
از نظر تظاهرات بالینی و شدت علائم نیز تفاوت چشمگیری و معناداری بین مردان و زنان گزارش نشده است. هر دو جنس در صورت عدم درمان، دچار علائم گوارشی، عصبی و چشمی مشابهی میشوند. پاسخ به درمان با ویتامینها و رژیم غذایی نیز در هر دو جنس یکسان است. تنها تفاوتهای احتمالی ممکن است مربوط به مسائل فیزیولوژیک طبیعی بین دو جنس باشد. به عنوان مثال، زنان مبتلا ممکن است در دوران قاعدگی به دلیل کمبود احتمالی ویتامین K و اختلالات انعقادی، خونریزیهای شدیدتری را تجربه کنند، یا در دوران بارداری با چالشهای خاصی روبرو شوند که مختص فیزیولوژی بارداری است، اما ماهیت خود بیماری و مکانیسم آسیبرسانی آن در مردان و زنان کاملاً یکسان عمل میکند.
مسائل مربوط به باروری نیز میتواند در هر دو جنس تحت تأثیر قرار گیرد اما اطلاعات در این زمینه محدود است. با توجه به نادر بودن بیماری، مطالعات گستردهای در مورد تفاوتهای ریز جنسیتی انجام نشده است، اما اصول کلی ژنتیک و پاتوفیزیولوژی حاکی از برابری کامل شرایط در هر دو جنس است. بنابراین، رویکردهای تشخیصی و درمانی فارغ از جنسیت بیمار پیگیری میشود و پروتکلهای درمانی استاندارد برای همه یکسان اجرا میگردد.
علت ابتلا به آبتالیپوپروتئینمی
علت اصلی و بنیادین ابتلا به آبتالیپوپروتئینمی، وجود یک جهش ژنتیکی در ژنی به نام MTTP است. این ژن دستورالعمل ساخت پروتئینی به نام “پروتئین انتقالدهنده تریگلیسیرید میکروزومال” (Microsomal Triglyceride Transfer Protein) یا همان پروتئین MTP را صادر میکند. برای درک اینکه چرا این پروتئین مهم است، باید به فرآیند هضم و انتقال چربی در بدن نگاه کنیم. چربیها و کلسترول برای حرکت در جریان خون (که پایه آبی دارد) نیاز دارند درون بستههایی پروتئینی قرار گیرند. پروتئین MTP مانند یک کارگر در کارخانههای سلولی (در روده و کبد) عمل میکند که وظیفهاش قرار دادن مولکولهای چربی درون پروتئینی به نام آپولیپوپروتئین B (ApoB) است تا ساختارهایی به نام شیلومیکرون (در روده) و VLDL (در کبد) تشکیل شوند.
در افراد مبتلا به آبتالیپوپروتئینمی، به دلیل نقص در ژن MTTP، پروتئین MTP یا اصلاً تولید نمیشود یا عملکرد ناقصی دارد. در نتیجه، سلولهای روده نمیتوانند چربیهای جذب شده از غذا را بستهبندی کرده و به صورت شیلومیکرون وارد جریان خون کنند. این باعث میشود چربیها در داخل سلولهای روده گیر بیفتند و در نهایت دفع شوند و هرگز به خون نرسند. به همین دلیل سطح چربی خون این افراد بسیار پایین است.
به طور مشابه، کبد نیز نمیتواند VLDL (که پیشساز LDL است) تولید کند. نبودن این حاملهای چربی (لیپوپروتئینهای حاوی ApoB) در خون، دو مشکل اصلی ایجاد میکند: اول اینکه بافتهای بدن سوخت و مواد سازنده غشایی کافی دریافت نمیکنند، و دوم اینکه ویتامینهای محلول در چربی (A, D, E, K) که برای جابجایی در خون مسافر همین شیلومیکرونها هستند، جذب نمیشوند. علت تغییر شکل گلبولهای قرمز (آکانتوسیتوز) نیز تغییر در ترکیب چربیهای غشای سلول به دلیل همین اختلالات متابولیک است. بنابراین، ریشه تمام مشکلات پیچیده این بیماری، نقص در عملکرد یک پروتئین واحد انتقالدهنده چربی است.
درمان دارویی آبتالیپوپروتئینمی
همانطور که پیشتر اشاره شد، دارویی برای اصلاح ژن معیوب وجود ندارد و “درمان دارویی” در واقع شامل تجویز تهاجمی و دقیق مکملها است. پزشکان از دوزهای فارماکولوژیک (بسیار بالاتر از دوز تغذیهای) استفاده میکنند.
ویتامین E: حیاتیترین دارو است. دوز معمول میتواند بین ۱۰۰ تا ۳۰۰ میلیگرم به ازای هر کیلوگرم وزن بدن در روز باشد. فرمهای محلول در آب ویتامین E (مانند توکوفرسولان) ممکن است جذب بهتری داشته باشند. هدف، رساندن سطح ویتامین E خون به محدوده طبیعی برای محافظت از سیستم عصبی است.
ویتامین A: برای سلامت چشم تجویز میشود. دوز آن باید با احتیاط تنظیم شود زیرا تجمع بیش از حد آن میتواند سمی باشد (اگرچه در این بیماران جذب پایین است).
ویتامین K: برای جلوگیری از اختلالات انعقادی تجویز میشود.
ویتامین D و کلسیم: برای سلامت استخوان.
گاهی اوقات مکملهای آهن یا اسید فولیک نیز در صورت وجود کمخونی تجویز میشوند. در مواردی که بیمار دچار مشکلات عصبی مانند درد نوروپاتیک یا اسپاسم عضلانی است، داروهای مربوطه توسط متخصص مغز و اعصاب برای کنترل علائم تجویز میگردد. هیچ داروی شیمیایی خاصی برای افزایش تولید MTP وجود ندارد و تحقیقات در زمینه ژندرمانی هنوز در مراحل اولیه و تحقیقاتی است.
درمان خانگی آبتالیپوپروتئینمی
درمان خانگی در واقع امتداد مدیریت پزشکی در محیط زندگی است. مهمترین بخش مراقبت در منزل، پایبندی دقیق به رژیم غذایی است. والدین و خود بیماران باید یاد بگیرند که برچسب مواد غذایی را با دقت بخوانند و از مصرف غذاهای حاوی چربیهای زنجیره بلند (LCT) خودداری کنند. آمادهسازی غذاها باید به روشهای بخارپز، آبپز یا کبابی بدون روغن باشد. استفاده از روغن MCT در پخت و پز یا اضافه کردن آن به سالاد، راهکاری خانگی برای تأمین انرژی است.
مراقبت از پوست نیز میتواند بخشی از درمان خانگی باشد. خشکی پوست ناشی از کمبود اسیدهای چرب ضروری و ویتامین A شایع است، بنابراین استفاده منظم از مرطوبکنندههای مناسب توصیه میشود. برای بیمارانی که دچار مشکلات تعادلی هستند، ایمنسازی محیط خانه (برداشتن فرشهای لغزنده، نصب دستگیره در حمام، نور کافی در شب) برای پیشگیری از سقوط و آسیب فیزیکی ضروری است. حمایت روانی در خانه، تشویق کودک به فعالیتهای متناسب با توانایی و ایجاد محیطی شاد و بدون استرس، تأثیر زیادی بر کیفیت زندگی دارد. همچنین خانواده باید نسبت به علائم هشداردهنده مانند تغییر در دید یا کبودیهای بیدلیل هوشیار باشند و سریعاً به پزشک مراجعه کنند.
رژیم غذایی مناسب برای آبتالیپوپروتئینمی
رژیم غذایی در آبتالیپوپروتئینمی یک انتخاب نیست، بلکه یک ضرورت حیاتی برای جلوگیری از علائم است. اصل کلی رژیم، محدودیت شدید چربیهاست. میزان مصرف چربی معمولاً باید کمتر از ۱۵ تا ۲۰ گرم در روز باشد.
غذاهای ممنوع: کره، خامه، روغنهای گیاهی معمولی (آفتابگردان، ذرت، زیتون)، گوشتهای پرچرب، فست فودها، مغزها (گردو، بادام) در مقادیر زیاد، زرده تخممرغ و شیر پرچرب. مصرف این مواد بلافاصله باعث درد شکم و اسهال میشود.
غذاهای مجاز: میوهها، سبزیجات، غلات کامل (برنج، نان، ماکارونی)، سفیده تخممرغ، گوشت سینه مرغ بدون پوست، ماهیهای کمچرب و لبنیات بدون چربی.
نقش روغن MCT: از آنجا که حذف چربی باعث کمبود کالری میشود، روغن تریگلیسیرید با زنجیره متوسط (MCT Oil) جایگزین میشود. این روغن از نارگیل یا هسته خرما استخراج میشود و ویژگی منحصر به فردی دارد که بدون نیاز به نمکهای صفراوی و شیلومیکرون جذب میشود. میتوان آن را به غذاها اضافه کرد، اما باید به تدریج وارد رژیم شود زیرا مصرف ناگهانی آن هم میتواند باعث دلدرد شود. همچنین مصرف مکملهای اسیدهای چرب ضروری (امگا ۳ و ۶) تحت نظارت پزشک ممکن است لازم باشد زیرا بدن قادر به جذب آنها از منابع طبیعی نیست.
عوارض و خطرات آبتالیپوپروتئینمی
اگر آبتالیپوپروتئینمی درمان نشود، عوارض آن ویرانگر خواهد بود. خطرناکترین عوارض مربوط به سیستم عصبی و چشم است.
نابینایی: رتینیت پیگمنتوزا درماننشده میتواند منجر به از دست دادن کامل بینایی در جوانی یا میانسالی شود.
ناتوانی حرکتی: آتاکسی پیشرونده و ضعف عضلانی میتواند بیمار را وابسته به صندلی چرخدار کند. نوروپاتی محیطی باعث از دست دادن حس در اندامها میشود که خطر زخم و عفونت را بالا میبرد.
مشکلات قلبی: فیبروز بافت قلب و کاردیومیوپاتی در برخی موارد گزارش شده است که میتواند منجر به نارسایی قلبی شود.
مشکلات خونی: کمبود ویتامین K میتواند منجر به اختلالات انعقادی و خونریزیهای داخلی خطرناک (مثلاً خونریزی مغزی) شود. کمخونی ناشی از همولیز (تخریب گلبولهای قرمز تغییر شکل یافته) نیز ممکن است رخ دهد.
مشکلات اسکلتی: پوکی استخوان و انحراف ستون فقرات ناشی از کمبود ویتامین D و ضعف عضلات حمایتکننده ستون فقرات، کیفیت زندگی را کاهش میدهد. تأخیر در رشد و بلوغ نیز از عوارض شایع در کودکان است. با این حال، با درمان مناسب و زودهنگام، بسیاری از این عوارض قابل پیشگیری یا کنترل هستند.
آبتالیپوپروتئینمی در کودکان و در دوران بارداری
در کودکان، بیماری معمولاً در ماههای اول تولد خود را نشان میدهد. چالش اصلی در کودکان، تأمین مواد مغذی کافی برای رشد (Growth) است. کودکی که چربی جذب نمیکند، کالری کافی برای وزنگیری و قد کشیدن ندارد. والدین با کودکی مواجه هستند که همیشه گرسنه است اما رشد نمیکند و شکمی متورم دارد. تشخیص دیرهنگام در این سنین میتواند باعث عقبماندگی رشد دائمی شود. همچنین کودکان در سنین مدرسه ممکن است به دلیل مشکلات تعادلی یا ضعف عضلانی در فعالیتهای ورزشی دچار مشکل شوند که نیازمند حمایت عاطفی و درک اولیای مدرسه است.
در دوران بارداری، زنان مبتلا به آبتالیپوپروتئینمی با چالشهای خاصی روبرو هستند. اولاً، باروری ممکن است طبیعی باشد اما نیاز به مراقبت دقیق دارد. مهمترین مسئله، تأمین ویتامینها و مواد مغذی برای جنین است. جنین برای رشد مغز و سیستم عصبی خود به کلسترول و اسیدهای چرب نیاز دارد. مادر مبتلا سطح کلسترول خون بسیار پایینی دارد، اما خوشبختانه بدن مکانیسمهای دیگری برای تأمین نیاز جنین دارد. با این حال، مکملیاری دقیق مادر حیاتی است. خطر خونریزی حین زایمان به دلیل کمبود ویتامین K مادر وجود دارد که باید قبل از زایمان با تزریق ویتامین K مدیریت شود. گزارشهای نادری از موفقیتآمیز بودن بارداری در این زنان وجود دارد، اما این بارداریها “پرخطر” محسوب شده و نیازمند پایش مداوم تیم تخصصی هستند.
مکانیسمهای آکانتوسیتوز و اثرات هماتولوژیک
یکی از جنبههای جذاب و در عین حال تشخیصی آبتالیپوپروتئینمی، تأثیر آن بر سلولهای خونی است. همانطور که گفته شد، گلبولهای قرمز در این بیماران به شکل “آکانتوسیت” یا سلولهای خاردار در میآیند. اما چرا این اتفاق میافتد؟ غشای گلبول قرمز از دو لایه چربی (لیپید) تشکیل شده است که تعادل دقیقی بین کلسترول و فسفولیپیدها در آن وجود دارد. در آبتالیپوپروتئینمی، به دلیل تغییرات شدید در لیپیدهای پلاسما، ترکیب چربیهای غشای گلبول قرمز نیز تغییر میکند. به طور خاص، نسبت اسفنگومیلین به لسیتین در غشا افزایش مییابد.
این تغییر شیمیایی باعث میشود غشای سلول سفتتر شده و انعطافپذیری خود را از دست بدهد. در نتیجه، وقتی این سلولها از مویرگهای باریک و طحال عبور میکنند، نمیتوانند شکل خود را به خوبی تغییر دهند و دچار تغییر شکلهای دائمی میخمانند میشوند. این سلولهای تغییر شکل یافته عمر کوتاهتری نسبت به گلبولهای قرمز سالم دارند و زودتر توسط طحال تخریب میشوند. این فرآیند میتواند منجر به نوعی کمخونی خفیف به نام “کمخونی همولیتیک” شود. علاوه بر این، وجود این سلولها در اسمیر خون، یک نشانه بسیار ارزان و در دسترس برای پزشکان است تا قبل از انجام آزمایشهای گرانقیمت ژنتیکی، به تشخیص صحیح نزدیک شوند. درک این مکانیسم نشان میدهد که چگونه یک نقص در متابولیسم چربی کبد، میتواند شکل ظاهری سلولهای خونی را تغییر دهد.
طول درمان آبتالیپوپروتئینمی چقدر است
آبتالیپوپروتئینمی یک وضعیت مادامالعمر است. ژن معیوب همیشه با بیمار خواهد بود و توانایی بدن برای تولید لیپوپروتئینهای حاوی ApoB هرگز به طور خودبهخود بازنمیگردد. بنابراین، طول درمان برابر با طول عمر بیمار است. هیچ “دورهی درمانی” مشخصی وجود ندارد که پس از آن بیمار بهبود یابد و بتواند داروها را قطع کند.
بیماران باید از نوزادی تا کهنسالی رژیم غذایی کمچرب را رعایت کنند و مکملهای ویتامینی را روزانه مصرف نمایند. قطع درمان، حتی برای دورههای کوتاه، میتواند باعث بازگشت سریع علائم گوارشی و شروع مجدد آسیبهای عصبی شود. خبر خوب این است که امید به زندگی (Life Expectancy) در بیمارانی که زود تشخیص داده میشوند و به درمان پایبند هستند، میتواند نزدیک به افراد عادی باشد. بسیاری از بیماران با مدیریت صحیح میتوانند تحصیل کنند، کار کنند و زندگی فعالی داشته باشند. با این حال، آنها نیازمند ویزیتهای منظم پزشکی (معمولاً هر ۶ تا ۱۲ ماه) برای بررسی سطح ویتامینها، معاینه چشم و بررسی وضعیت کبد (برای بررسی کبد چرب که گاهی رخ میدهد) هستند. پایداری در درمان، کلید داشتن یک زندگی طولانی و با کیفیت است.
جمعبندی
آبتالیپوپروتئینمی یا سندرم باسن-کورنزوایگ، یک اختلال ژنتیکی نادر است که در آن بدن توانایی تولید شیلومیکرون و VLDL را به دلیل نقص در ژن MTTP از دست میدهد. این امر منجر به سوءجذب شدید چربیها و ویتامینهای محلول در چربی (A, D, E, K) میشود. علائم اصلی شامل اسهال چرب، عدم رشد، مشکلات تعادلی (آتاکسی)، رتینیت پیگمنتوزا و وجود گلبولهای قرمز خاردار (آکانتوسیت) در خون است. اگرچه درمان قطعی وجود ندارد، اما با تشخیص زودهنگام و مدیریت دقیق شامل رژیم غذایی بسیار کمچرب، استفاده از روغن MCT و مصرف دوزهای بسیار بالای ویتامین E و سایر ویتامینها، میتوان از عوارض شدید عصبی و نابینایی جلوگیری کرد و کیفیت زندگی بیمار را حفظ نمود. درمان این بیماری نیازمند تعهد مادامالعمر به رژیم دارویی و غذایی است.